SUBMIT YOUR MANUSCRIPT
Affiliations
ABSTRACT
Transfusion-associated graft versus host disease (TA-GvHD) is a rare complication of blood transfusion. It is a delayed transfusion reaction that usually presents from day 7 to day 14 following a transfusion. It can affect both immunocompromised as well as immunocompetent patients. In immunocompetent patients, the culprit is usually the transfusion from first-degree relatives. The recipient takes the donor cells as self while the donor takes the recipient cells as foreign and the lymphocytes in the donor blood start attacking the recipient cells leading to a cascade of catastrophic events that ultimately result in a condition called TA-GvHD. The condition is usually fatal in ≥90% of cases. Here, we present one such case in which the maternal blood transfused to an infant cost him his life within 12 days of transfusion.
Key Words: Transfusion, Homozygous antigen, Heterozygous antigen, Graft versus host disease.
INTRODUCTION
Transfusion-associated graft versus host disease (TA-GvHD) is a rare complication of blood transfusion leading to death in ≥90% of cases. It is due to an immunologic attack by the viable donor lymphocytes contained in the transfused blood component against the transfusion recipient. In immunocompetent patients, donor lymphocytes initiate an immune response in the recipient causing damage to tissues.1 Patients present with symptoms such as fever, watery diarrhoea (may be accompanied by abdominal pain and bloody stools), maculopapular rash (usually starts centrally and then spreads to peripheral regions), liver injury evident by elevated liver enzymes and bone marrow aplasia causing pancytopenia after 3 to 30 days of transfusion of non-irradiated cellular blood component. The inability of the host to recognise and remove donor lymphocytes either due to immune defects in immunocompromised patients or donor homozygosity for an HLA haplotype for which the recipient was heterozygous in immunocompetent patients results in TA-GVHD.2
A history of transfusion from first-degree relatives, specific signs and symptoms, and a biopsy (tissue or hepatic) is required for the diagnosis of TA-GVHD.1 The definitive diagnosis requires the demonstration of leukocyte chimerism by detection of donor lymphocytes in the recipient blood.
The pathogenesis of the disease involves the existence of three conditions in a recipient to develop TA-GVHD: HLA antigen difference between donor and recipient, presence of viable donor immunocompetent cells in the blood component, and a recipient incapable of rejecting the donor immunocompetent cells.3 At-risk immunodeficient patient populations include infants and patients with cancer or compromised immune systems. In patients with an intact immune system, the TA-GVHD can occur when the patient is transfused with a cellular blood component from a donor homozygous for an HLA haplotype that is shared with the heterozygous recipient. The transfused lymphocytes are able to recognise the recipient’s non-shared haplotype as foreign and will mount an immune attack on the recipient.2
Here, we present one such case in which the maternal blood transfused to an infant cost him his life within 12 days of transfusion.
CASE REPORT
A 10-month male born from consanguineous marriage in a village in a far-flung area of south Punjab, had been suffering from off-and-on fever and recurrent diarrhoea for the last four months. He was taken to multiple general practitioners who placed him on multiple antipyretics and antibiotics. The child started developing increasing pallor associated with lethargy and decreased appetite. The child was taken to a paediatrician who started him on oral hematinics. The hematinics were continued for 15 days but no improvement was noted. His blood complete picture revealed low haemoglobin (8 g/dl). The paediatrician advised the child blood transfusion.
The child was transfused 100 ml of whole blood from the mother. The child remained in his usual state of health for one week; when he developed an erythematous rash on his abdomen sparing the extremities (Figure 1). He was treated symptomatically as a case of chicken pox but his condition worsened. The local paediatrician referred him to a teaching hospital for further management. In the meanwhile, he developed profuse diarrhoea. Considering the history, a differential diagnosis of complications of chicken pox and liver failure was made. His blood tests revealed a drastic drop in haemoglobin, total leukocyte count (TLC), and platelet counts. His liver function tests revealed elevated alanine transaminase (ALT) and bilirubin.
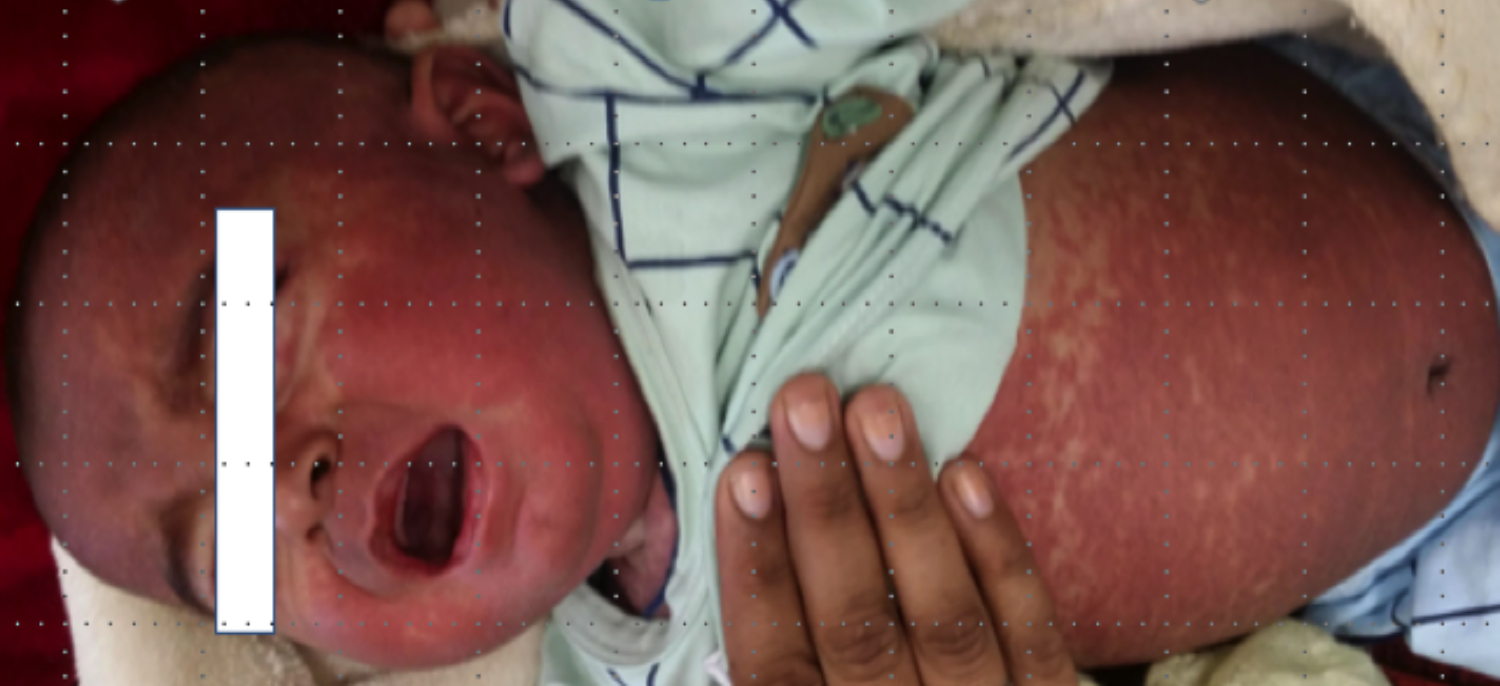 Figure 1: Image showing patient’s erythema and rash.
Figure 1: Image showing patient’s erythema and rash.
Due to a lack of proper facilities at the peripheral teaching hospital, the child was referred to our setup. On arrival, the child was semiconscious, having a temperature of 101˚F, pulse of 140 beats/min, and respiratory rate of 35 breaths/min. On examination, he was pale having a red-coloured macular rash on his abdomen and face, sparing extremities. He had yellowish discolouration of the sclera but no cyanosis, lymphadenopathy, and organomegaly.
Considering the history of transfusion of maternal blood and the development of diarrhoea, deranged liver function tests, and a rash sparing the extremities, a diagnosis of TA-GVHD was made. Without indulgence in diagnostic modalities, the child was rushed to an inpatient facility where he was started on immunosuppressant therapy (injection corticosteroid and cyclosporine). The preliminary tests were repeated which showed pancytopenia (haemoglobin, 4 g/dl , TLC, 1.2×109/l, platelet count, 50×109/l) and increased hepatic enzyme levels (serum bilirubin, 40 mmol/l (Normal: 10-20 mmol/l), serum alanine aminotransferase (ALT), 140 IU/l (Normal: 10-40 IU/l), serum aspartate aminotransferase (AST), 128 IU/l (Normal: 10-40 IU/l), and serum gamma glutamyltransferase, 80 IU/l (Normal: 5-55IU/l) confirming the suspicion of TA-GvHD.
Unfortunately, the child could not survive despite the efforts to control the disease. The confirmatory tests to determine donor chimerism (human leukocyte antigens (HLA) matching and short tandem repeats [STR] analysis) could not be done due to the non-affordability of attendants of patient and the demise of the child.
DISCUSSION
In 1966, the first case of TA-GvHD was reported in Japan. The Japanese population has a high risk of developing TA-GvHD (1 in 874 transfusions from a random donor), resulting from the closed island’s reduced genetic diversity and HLA homogeneity. The development of TA-GvHD depends on the immune response of the host, the characteristics of the components transfused, and the genetic diversity of donor-recipient HLA relationship. TA-GvHD has been associated with whole blood and red cell concentrate (RCC) transfusions but never with transfusions of fresh frozen plasma (FFP) and cryoprecipitate that lack viable lymphocytes.1
The disease is usually initially suspected on suggestive transfusion history as symptoms typically present within 1 to 3 weeks, post-transfusion. Clinical correlation of the history and the clinical signs and symptoms affecting the skin, gastrointestinal system, liver, and bone marrow is usually required for accurate and rapid diagnosis.2 The differential diagnosis usually includes viral infections, liver failure, and drug reactions.
Unfortunately, in this patient’s case, the disease was initially mismanaged by being treated as chickenpox, highlighting the lack of knowledge about safe blood transfusion practices in peripheral settings of our country.3,4 The available literature contains only two case reports from Pakistan.5,6 The paucity of available data does not indicate that TA-GvHD is a very rare complication, but rather it is underdiagnosed and underreported.
For confirmation of the diagnosis of TA-GvHD, it is necessary to detect the presence of donor lymphocytes in the blood of the recipient, indicating leukocyte chimerism. The patients who are at risk of developing TA-GvHD include those with compromised immune systems, such as cancer patients or infants like the present patient.2 Even patients with intact immune systems can develop TA-GvHD if they receive a cellular blood component from a donor who shares an HLA haplotype with the recipient but is homozygous for it. In this case, the recipient’s immune system recognises the donor lymphocytes as foreign and mounts an immune attack on them, whereas the transfused lymphocytes recognise the recipient’s non-shared haplotype as foreign and attack the recipient’s own cells.3
The only method of prevention of TA-GvHD is via leuko-reduction, achieved through irradiation of blood products, which can prevent TA-GvHD and has successfully reduced the incidence of this complication in the UK, but it is a facility not commonly available in Pakistan. The culture of consanguineous marriages in our population (a major cause of reduced genetic diversity) and the lack of awareness regarding safe blood transfusion practices increase the likelihood of a relatively restricted gene pool and incidence of TA-GvHD.
In conclusion, TA-GvHD is a rare but potentially fatal complication of blood transfusions, which is often underdiagnosed and underreported. To prevent future cases of TA-GvHD, it is important to increase awareness about safe blood transfusion from the first-degree relatives and the use of irradiated blood products when necessary. This can be achieved through education campaigns at both individual and community levels.
PATIENT’S CONSENT:
Informed consent was taken from the patient's parents to publish this case.
COMPETING INTEREST:
The authors declared no competing interest.
AUTHORS’ CONTRIBUTION:
SF, AM, NS, RAKL, TG: Substantial contribution to the conception or design of the work, acquisition, analysis or interpretation of data for the work, drafting the work and revising it critically for important intellectual content.
REFERENCES